23.01.2018
ALS: Dresdner Grundlagenforscher entdecken neuen Krankheitsmechanismus bei Amyotropher Lateralsklerose

Marcel Naumann, Dr. Arun Pal und Prof. Dr. Dr. Andreas Hermann diskutieren die Ergebnisse ihrer Studie.
Das Dresdner Forschungsteam um Prof. Dr. Dr. Andreas Hermann aus dem Bereich Neurodegenerative Erkrankungen der Klinik für Neurologie am Universitätsklinikum Carl Gustav Carus hat in grundlagenwissenschaftlichen Arbeiten an menschlichen Nervenzellen von Patienten mit der Erkrankung Amyotrophe Lateralsklerose (ALS) einen neuen Krankheitsmechanismus entdeckt. Bisher wurde als wesentlicher, krankheitsverursachender Mechanismus dieser Erkrankung oft die krankhafte Ablagerung fehlgefalteter Proteine (Eiweiße) angesehen. Die Arbeitsgruppe zeigt nun, dass Fehlfunktionen des zellulären Reparatursystems für Schäden im Erbgut (DNA) der Zelle zeitlich vor den Proteinablagerungen beobachtet und ursächlich miteinander verknüpft werden konnten. Das eröffnet völlig neue Forschungs- und Behandlungsperspektiven für ALS. Ihre Erkenntnisse veröffentlichten die Wissenschaftler jetzt im Fachjournal Nature Communications (DOI: 10.1038/s41467-017-02299-1) in einer mit zahlreichen anderen nationalen Kooperationspartnern – insbesondere den Universitätskliniken Ulm und Aachen – erarbeiteten Studie. Mögliche Therapeutika gegen diese beobachteten Fehlfunktionen des zellulären Reparatursystems sind in Phase 1-Studien bei Tumorerkrankungen, wo sie schon länger als Ursache bekannt sind, bereits in Erprobung.
Amyotrophe Lateralsklerose (ALS) ist eine neurodegenerative Erkrankung, bei der es zum unaufhaltsamen Untergang sämtlicher motorischer Nervenzellen kommt. Die Patienten leiden unter zunehmenden Lähmungen aller willkürlich innervierbaren Muskeln, was innerhalb von zwei bis fünf Jahren zum Tod führt. Die Erkrankung ist nicht heilbar, die einzig zugelassenen Therapien verlängern das Überleben um lediglich wenige Monate.
Unter der Verwendung von humanen, induzierten pluripotenten Stammzellen (hiPSZ), die aktuell als modernstes Modellsystem für die Erforschung neurodegenerativer Erkrankungen angesehen werden, gelangte die Dresdner Arbeitsgruppe um Prof. Dr. Dr. Andreas Hermann zur Erkenntnis, dass es bei der sogenannten FUS-ALS zu einem fortschreitenden Untergang des motorischen Nervenausläufers (peripher-motorischen Axons) kommt, gefolgt vom Zelltod und der pathologischen Ablagerung fehlgefalteter Proteine. Weiterhin wiesen die motorischen Nervenzellen deutliche Defizite bei der Reparatur von DNA-Schäden im Erbgut auf, einem Mechanismus, der insbesondere in der Tumorbiologie eine große Rolle spielt.
Klinisch besonders relevant ist die Schlüsselbeobachtung, dass die gestörte Reparatur von DNA-Schäden als wesentlicher initialer Auslöser für Neurodegeneration und pathologische Proteinablagerungen identifiziert werden konnte: wenn immer in den Versuchen die Maschinerie der DNA-Schadensreparatur korrigiert wurde, kam es auch zu einer vollkommenen Genesung der Neurodegeneration und pathologischen Proteinablagerungen. Diese krankhaften Veränderungen konnte abschließend in menschlichem Hirn- und Rückenmarksmaterial von ALS-Patienten bestätigt werden. Dabei wurden Substanzen entdeckt bzw. für wirksam befunden, die bereits seit langem in der Tumortherapie diskutiert werden und für die es bereits erste Phase 1-Studien bei Tumorpatienten gibt.
„Eine Ankopplung an diese zu relevanten Wirkstoffen in anderem Zusammenhang bereits laufenden Studien würde die rasche Umsetzung in eine klinische Anwendung für ALS-Patienten deutlich früher ermöglichen, als wenn man komplett durch die normale Phasenentwicklung von Pharmaka gehen müsste“, sagt Prof. Dr. Dr. Andreas Hermann, Leiter der Studie. Dennoch rechnet er noch mit einigen weiteren Jahren Arbeit, bevor dies Patienten zugutekommen könnte.
„Diese Ergebnisse sind deshalb so relevant, weil nach gängiger Lehrmeinung bei den meisten neurodegenerativen Erkrankungen die Ablagerung pathologisch gefalteter Proteine als wesentlicher Krankheitsmechanismus angesehen wird. Folglich zielen die meisten aktuellen Therapieansätze auf die Beseitigung oder Verringerung dieser fehlgefalteten Proteine ab“, so Prof. Hermann weiter. „Die Ergebnisse unserer Studie legen aber die Notwendigkeit ganz andersartiger Therapien nahe, die viel gezielter die tatsächliche, grundlegende Pathophysiologie behandeln und die pathologische Ablagerung von Proteinen an der Wurzel packen würden“, sagt Marcel Naumann, Erstautor dieser Studie.
„Dass die Reparatur von DNA-Schäden bei neurodegenerativen Erkrankungen gestört sein kann, ist nicht neu. Eine derart zentrale Schlüsselrolle dieses Mechanismus in der Pathologie einer neurodegenerativen Erkrankung hat uns jedoch überrascht und stellt eine der wesentlichen Erkenntnisse dieser Studie dar“, so Dr. Arun Pal, ebenfalls Erstautor der Studie. „Dies eröffnet ganz neue Forschungsperspektiven, bei denen die gemeinsamen Mechanismen der Tumorbiologie und Biologie neurodegenerativer Erkrankungen in den Mittelpunkt rücken“, so Prof. Hermann.
Es ist nun geplant, ein entsprechendes Konsortium aus Tumorbiologen und Neurologen ins Leben zu rufen, um diese Mechanismen im Detail weiter zu untersuchen.
„Die hier vorgelegte Arbeit demonstriert eindrucksvoll die wissenschaftliche Stärke der Dresdner Hochschulmedizin, aber auch des gesamten Dresdner Forschungsstandortes, da sie in enger Kooperation zwischen Arbeitsgruppen der Klinik und Poliklinik für Neurologie, der Klinik und Poliklinik für Nuklearmedizin, des Forschungsverbundes OncoRay – wie aber auch des Max-Planck-Instituts für Zellbiologie und Genetik Dresden, des Deutschen Zentrums für Neurodegenerative Erkrankungen Dresden und des DFG-Centers for Regenerative Therapies Dresden entstand“, sagt Prof. Dr. Michael Albrecht, Medizinischer Vorstand des Universitätsklinikums Dresden. „Diese Zusammenarbeit der Forscher universitärer und außeruniversitärer Einrichtungen ist ein hervorragendes Beispiel für den Gedanken von „DRESDEN-concept“, dem Verbund der TU Dresden mit starken Partnern aus Wissenschaft und Kultur mit dem Ziel, die Exzellenz der Dresdener Forschung sichtbar zu machen“, unterstreicht Prof. Dr. Heinz Reichmann, Dekan der Medizinischen Fakultät Carl Gustav Carus der TU Dresden und Direktor der Klinik und Poliklinik für Neurologie des Universitätsklinikums Dresden.
Das Forschungsvorhaben wurde unter anderem gefördert vom virtuellen Helmholtz-Institut “RNA dysmetabolism in ALS and FTD (VH-VI-510)”, dem Else-Kröner-Promotions-Kolleg Dresden, dem Deutschen Zentrum für Neurodegenerative Erkrankungen DZNE, dem DFG-Center for Regenerative Therapies Dresden (CRTD), der Deutschen Gesellschaft für Muskelerkrankungen, der Roland Ernst Stiftung Sachsen und der NOMIS Stiftung sowie durch eine große Einzelspende einer Familie einer verstorbenen ALS Patientin.
Publikation:
Marcel Naumann, Arun Pal and Andreas Hermann: “Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation”; in: NATURE COMMUNICATIONS, DOI: 10.1038/s41467-017-02299-1, direct link http://rdcu.be/FmAs, www.nature.com/naturecommunications
Bild 1: Marcel Naumann, Dr. Arun Pal und Prof. Dr. Dr. Andreas Hermann diskutieren die Ergebnisse ihrer Studie (Foto: Stephan Wiegand, MF TU Dresden).
Bild 2: Schema zur aktuellen Studie – ausführliche Legende siehe unten *, Quelle: Naumann, Pal, Hermann, UKD.
Kontakt
Prof. Dr. med. Dr. rer. med. Andreas Hermann
Bereichsleiter
Bereich Neurodegenerative Erkrankungen
Klinik und Poliklinik für Neurologie
Universitätsklinikum Carl Gustav Carus Dresden
Fetscherstrasse 74
01307 Dresden, Germany
Phone: ++49-351-458-2532
Fax: ++49-351-458-4352
email:
Homepage: http://www.als-dd.de
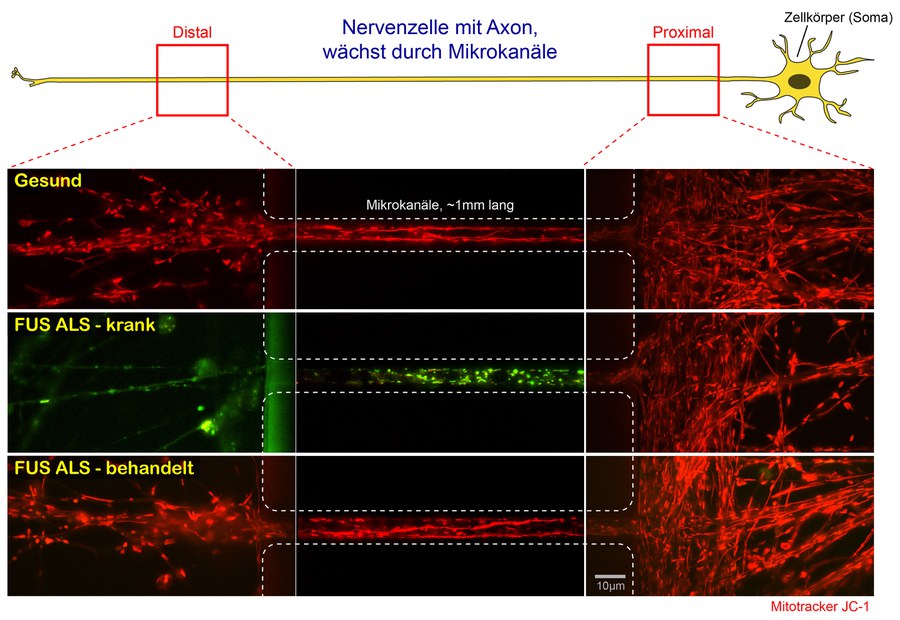
Schema zur aktuellen Studie – ausführliche Legende siehe unten *.
* Legende zum Schema „ALS-Forschung_Naumann-Pal-Hermann_Jan2018“
Nervenzellen von Patienten bilden in Kultur Fortsätze, sogenannte Dendriten und Axone. Die Axone wachsen proximal (rechts) in Mikrokanäle hinein und sprießen am distalen Ende (links) wieder heraus. Im Dunkelfeld des Videomikroskops können nun durch den speziellen Fluoreszenzfarbstoff JC-1 die Kraftwerke der Zelle, die sogenannten Mitochondrien, sichtbar gemacht und ihr axonaler Transport durch die Mikrokanäle verfolgt werden. Durch das Übereinanderlagern aller Einzelaufnahmen des Videos werden die Bewegungen der Mitochondrien durch langgezogene rote Bahnenlinien dargestellt (sogenannte maximum intensity projection).
Zusätzlich zeigt die Farbe von JC-1 an, ob die Membrane der Mitochondrien elektrisch polarisiert sind und somit Energie in Form von ATP produzieren können oder nicht (durch die sogenannte oxidative Phosphorylierung in der Atmungskette). Hierbei bedeutet rot: physiologische Polarisation und ATP Produktion, intakte Energieversorgung. Grün dagegen: keine Polarisation und kein ATP, lokale Energieengpässe.
Der obere Kanal zeigt gesunde Nervenzellen, in der alle Mitochondrien durchgängig ein intaktes Membranpotential (rot) und rege Bewegungen aufweisen.
Der mittlere Kanal dagegen zeigt Nervenzellen eines FUS-ALS Patienten. Deutlich erkennt man den Verlust des Membranpotentials (rot, noch intakt) und der Bewegung (rote Bahnen) von proximal (rechts) nach distal (links) durch den Umschlag nach grün (Stillstand, kaum noch Bahnen).
Der untere Kanal zeigt die gleichen FUS-ALS Nervenzellen, die in Kultur mit einer vielversprechenden Substanz behandelt wurden, die die gestörte DNA Schadensreparatur im Zellkern wieder herstellt. Deutlich zu erkennen sind die Restauration des mitochondrialen Membranpotentials (wieder durchgängig rot) und der Bewegungsprozesse (rote Bahnen).