T2: Force field simulation of guest induced gating phenomena
Rochus Schmid
The molecular understanding of guest molecule adsorption induced transformations of flexible MOFs necessitates theoretical simulations of large systems with many soft degrees of freedom. The key target of this project is to develop accurate force field based molecular models, based on the insight gained within T1 on the basis of electronic structure methods, but on a substantially larger length and timescale. In addition, a novel methodology for the simulation of flexible switchable MOFs in contact with a reservoir of guest molecules (osmotic ensemble), namely Grand Canonical Molecular Dynamics (GCMD), will be established and applied, using these force fields. In line with projects S1 and S2, force fields for pillared layer MOFs with different metal nodes, ranging from Ni, with its extreme breathing, up to more stiff Zn and Cu systems will be parameterized in a systematic way using QM reference data from T1. In addition, flexible side chains on the linkers, which allow an alternative access to flexibility, will be included into the parameterization. Linking with P1 and P2, a primary focus of the application calculations is on applying the GCMD method to help interpreting the in situ spectroscopic measurements of MOF transformations. The GCMD simulations can map the free energy of the system with respect to the guest molecule loading and a number of structural parameters, representing the transformation of the flexible MOF matrix (e.g. a lattice parameter). Such a map allows a complete interpretation of gating and breathing phenomena, but currently has been computed only under severe constraints or for simplified systems. By combining accurate atomistic force fields and meta-dynamics enhanced GCMD sampling it will be possible to compute such maps for the real systems of interest.
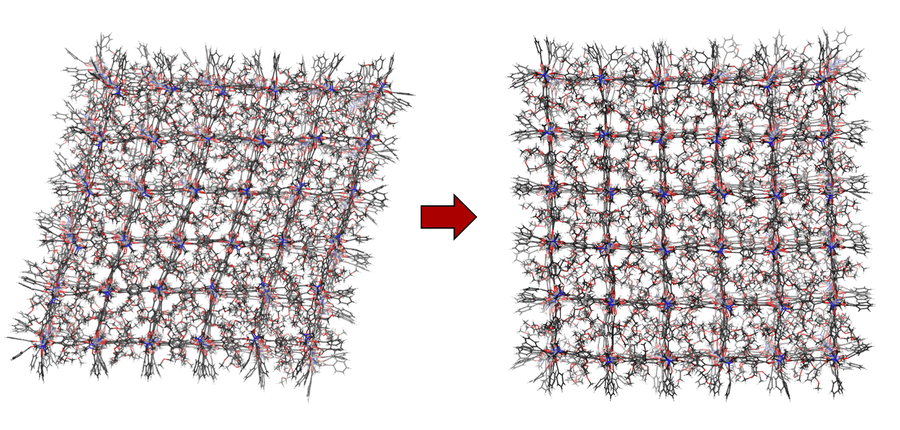
Fig.1: Opening of BME-fu-MOF nanoparticle of size 6x6x6 by heating from 300K to 500K.
J. P. Dürholt, R. Galvelis, R. Schmid, “Coarse graining of force fields for metal-organic frameworks“, Dalton. Trans. 2016, 45, 4370-4379.
M. Alaghemandi, R. Schmid, “A Model Study of Thermoresponsive Behavior of Metal-Organic Frameworks Modulated by Linker Functionalization“, J. Phys. Chem. C 2016, 120, 6835-6841.
R. Schmid, “An Electric Field Induced Breath for Metal-Organic Frameworks“, ACS Cent. Sci. 2017, 3, 369–371.
J. Keupp, R. Schmid, “TopoFF : MOF structure prediction using specifically optimized blue prints“, Faraday Discuss. 2018, 211, 79-101.
S. Impeng, R. Cedeno, J. P. Dürholt, R. Schmid, S. Bureekaew, “Computational structure prediction of 4,4-connected copper paddle-wheel-based MOFs: Influence of ligand functionalization on the topological preference“, Cryst. Growth Des. 2018, 18, 2699-2706.
J. P. Dürholt, G. Fraux, F.-X. Coudert, R. Schmid, “Ab initio derived force fields for Zeolitic Imidazolate Frameworks: MOF-FF for ZIF“, J. Chem. Theory Comput. 2019, DOI: 10.1021/acs.jctc.8b01041.