AG Klapproth

Wir sind ein junges, interdisziplinäres und diverses Team am Institut für Pharmakologie und Toxikologie der Medizinischen Fakultät der TU Dresden. Unser zentrales Forschungsinteresse liegt in der Aufklärung der komplexen Immunreaktionen nach Myokardinfarkt sowie in der Entwicklung nachhaltiger biopharmazeutischer Produktionsplattformen für innovative Therapeutika.
Immunpharmakologische Forschung nach Myokardinfarkt
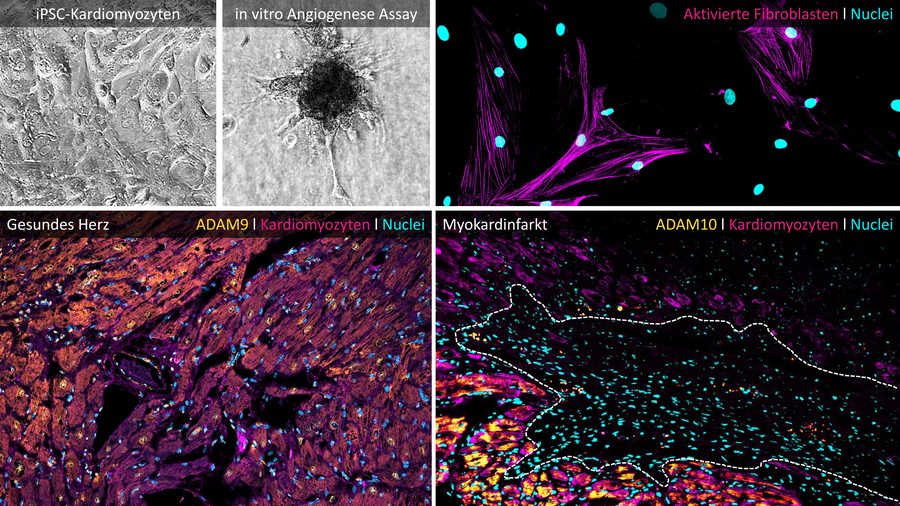
Nach einem Myokardinfarkt bestimmen frühe Immunprozesse nicht nur das Ausmaß der Entzündung, sondern legen die strukturelle und funktionelle Zukunft des Herzens fest. Unsere Forschung setzt genau an diesem kritischen Punkt an: Wir untersuchen, wie proteasegesteuerte Programme in Neutrophilen das kardiale Remodeling nach Infarkt aktiv formen – und wie sich diese Prozesse gezielt therapeutisch beeinflussen lassen.
Im Zentrum unserer Arbeit steht die funktionelle Heterogenität von Neutrophilen im infarzierte Herzen. Entgegen der klassischen Vorstellung als kurzlebige, rein proinflammatorische Effektorzellen zeigen unsere Daten, dass Neutrophilen in unterschiedliche Subpopulationen differenzieren, die über ihre Interaktion mit der extrazellulären Matrix, über Zell-Zell-Kommunikation und über gewebsdestruktive Programme maßgeblich zum strukturellen Umbau des Myokards beitragen. Diese bislang unterschätzten Neutrophilen-Subtypen stellen einen zentralen Treiber des postinfarktiven Remodelings dar.
Ein besonderer Fokus liegt dabei auf ADAM-Metalloproteasen, insbesondere ADAM9 und ADAM10, sowie der CX3CL1/CX3CR1-Achse als molekulare Schaltstellen der Immunzellrekrutierung, -aktivierung und -funktion. Wir verstehen Proteasen dabei nicht als passive Marker, sondern als aktive Regulatoren von Neutrophilen-Identität und -Funktion im Gewebe. Durch zelltypspezifisches Protease-Targeting eröffnen sich neue Möglichkeiten, entzündliche und gewebsumbauende Prozesse im Herzen präzise zu modulieren, ohne die notwendige Immunantwort global zu unterdrücken.
Methodisch kombinieren wir genetische Mausmodelle (einschließlich konditionaler Knock-outs), iPSC-abgeleitete humane Zellsysteme, CRISPR/Cas-basierte Genomeditierung, Multi-Omics-Ansätze (RNA-Seq, Proteomik, Secretom-Analysen), hochdimensionale Durchflusszytometrie, moderne Bildgebung sowie funktionelle kardiovaskuläre In-vivo-Modelle. Dieser integrative Ansatz erlaubt es uns, proteasegesteuerte Immunmechanismen über molekulare, zelluläre und funktionelle Ebenen hinweg systematisch zu entschlüsseln und in konkrete therapeutische Strategien zu überführen.
GreenPharming – Nachhaltige Produktion therapeutischer Proteine
Parallel verantwortet unsere Arbeitsgruppe einen Großteil des institutsweiten GreenPharming-Projekts. Ziel ist es, pflanzenbasierte (tierfreie) Produktionssysteme für biopharmazeutische Wirkstoffe wie Antikörper, Peptide und Zytokine zu etablieren. Diese sollen perspektivisch unter GMP-Bedingungen in einer zirkulären Bioökonomie in der Lausitz hergestellt werden.
Wir nutzen Pflanzen als Expressionsplattformen, etwa durch transienten Gentransfer in Nicotiana benthamiana, und entwickeln modulare Vektorsysteme, um eine schnelle und skalierbare Produktion zu ermöglichen. Die aufgereinigten Proteine werden umfassend hinsichtlich Struktur, Glykolisierung und Funktion charakterisiert (u. a. mittels Massenspektrometrie, HPLC, funktioneller Zellassays). Ein besonderer Fokus liegt auf innovativen Zytokinen für die Expansion von CAR-T- und CAR-NK-Zellen zur Anwendung in der personalisierten Zelltherapie.
Das Projekt verbindet biotechnologische Innovation mit Nachhaltigkeit: Restpflanzen sollen in Kreisläufen weiterverwertet, erneuerbare Energiequellen genutzt und regionale Wertschöpfungsketten in der Lausitz aufgebaut werden.

Unser Team
Unser Team vereint Nachwuchswissenschaftler:innen, Postdocs und technische Mitarbeitende aus unterschiedlichen Fachrichtungen wie Biologie, Biotechnologie, Medizin und Pharmazie. Wir arbeiten kollaborativ, kreativ und lösungsorientiert – interdisziplinär innerhalb der Fakultät, in enger Kooperation mit Industrie- und Forschungspartnern in der Region sowie international vernetzt.
Mit unserer Forschung möchten wir sowohl grundlegende biologische Mechanismen entschlüsseln als auch konkrete, nachhaltige und innovative Therapiekonzepte entwickeln, die langfristig den Weg in die klinische Anwendung finden können.

v.l.n.r.: Leo, Robby, Muazzez, Johanna, Erik, Marie, Nadine, Rizka, Patrick