Recombineering Guide
Attention:
Content is not getting updated any longer. For the newest developments in recombineering please see our recent publications.
Recombineering is a revolutionary method for DNA engineering using homologous recombination in E.coli. Recombineering allows unlimited cloning, subcloning, and modification of DNA at any chosen position. It permits precise engineering of DNA molecules of any size, including very large ones such as BACs or the E.coli chromosome.
Advantages over conventional methods:
- Independent of restriction sites
- No size limits
- No unwanted mutations
- Rapid
Here we present some important rules for successful recombineering.
How to get Recombineering Proficient Cells
-
Transform the expression plasmid pSC101-BAD-γβαAtet into the E. coli strain, in which you want to perform recombineering. [To prepare DNA of the expression plasmid, using any commercial plasmid preparation kit will result in very low yield due to the low copy number of the pSC101 plasmid. Please use our plasmid preparation protocol. To check your prepared expression plasmid rather perform a function test than a digestion analysis.]
-
To start overnight cultures, pick colonies from the respective plate.
-
Inoculate 1.5 ml reaction tubes containing 1.0 ml LB medium plus the appropriate antibiotics (select for BAC or plasmid and for expression plasmid).
-
Puncture the cap for aeration.
-
Incubate the cultures with shaking at 30 °C and 950 rpm o/n.
Next day
-
Set up punctured reaction tubes containing 1.4 ml fresh LB medium supplemented with the same antibiotics as in the o/n culture.
-
Inoculate each with 30 µl fresh overnight culture.
-
Incubate the tubes at 30 °C, 950 rpm for 2 hours.
Induction
-
Add 20 µl 10 % L-Arabinose to a final concentration of 0.1 %-0.2%. This will induce the expression of the proteins necessary for recombineering.
-
Leave some tubes without induction as negative controls.
-
Incubate at 37 °C, 950 rpm for 40 min.
L-arabinose stock solution
-
Use 10 % L-arabinose in dH2O, fresh or frozen in small aliquots at -20 °C
-
Use 20 μl stock solution per 1.4 ml LB for induction of recombination protein expression from pSC101-BAD-γβαAtet.
How to Make Cells Competent for Electroporation
Before starting
-
Chill dH2O/ 10% glycerol on ice for at least 2 hours.
-
Chill 1 mm gap electroporation cuvettes.
-
Refridgerate a cooling centrifuge to 2 ºC. (or work in a 4 ºC cold room with a normal centrifuge)
-
Use your fresh E. coli culture.
-
Put the cells on ice (and keep them on ice whenever possible).
-
Spin the cells down at 9,000 rpm (~ 7,800 rcf) for 30 sec in the cooling centrifuge at 2 °C.
-
Discard the supernatant by decanting. Discard as much supernatant as possible.
-
Resuspend the pellet in 1 ml of ice cold water/ 10% glycerol by pipetting.
-
Spin the cells down at 10,000 rpm (~ 9,600 rcf) for 30 sec in the cooling centrifuge at 2 °C.
-
Discard the supernatant by decanting. Discard as much supernatant as possible.
-
Resuspend the pellet in 1 ml of ice cold water/ 10% glycerol by pipetting.
-
Spin the cells down at 11,000 rpm (~ 11,600 rcf) for 30 sec in the cooling centrifuge at 2 °C.
-
Discard the supernatant by decanting and leave about 30 µl
-
Use the cells immediately
-
How to Electroporate
-
Add DNA (PCR product 100-1000 µg, or plasmid DNA plus PCR mix up to 1 µg) to the resuspended pellet in the reaction tube (see ProtocolI V.5.) and pipette the mixture into the chilled 1 mm electroporation cuvette.
-
Set the electroporator to 1350 V, 10 mF, 600 Ohms. (This setting belongs to an Eppendorf® Electroporator 2510 using an electroporation cuvette with a gap of 1 mm. Other devices can be used, but the voltage has to be fixed at 1350V and the length of the pulse should be 5 ms.)
-
Carefully knock the cuvette on the table to remove air bubbles and dry the metallic sides of the cuvette with a tissue. Do not touch the metallic sides with your hands.
-
Place the cuvette into the holder of the electroporator, insert it and push 2 times the pulse button.
-
Add 1 ml LB medium without antibiotics to the cuvette. Resuspend the cells carefully by pipetting up and down and pipette back into the reaction tube. (avoid air bubbles in the suspension)
-
Incubate the cultures at 37 °C with shaking at 950 rpm for 70 min.
-
Plate 30-100 µl for simple transformations or high efficieny recombineering (high copy plasmid modification or oligo repair step of counter selection). Plate 1 ml for recombineering with low efficiency (BAC modification). To plate 1 ml, spin down the tubes (e.g. 1 min, 11,000 g), discard the supernatant but leave about 50-100 µl, resuspend the pellet in the remaining liquid.
-
Streak on the plate in a way of dilution to ensure emerging of single colonies. Select for transformants or recombinants with corresponding antibiotics
-
Incubate at correct temperature (in case of pSC101 plasmid, at 30 °C)
To target your BAC/genome/plasmid at the site(s) you choose, the homologous region must be included in the two oligonucleotides for amplification of a drug selectable cassette. Therefore each oligonucleotide consists of two (or, if desired, three) parts:
- Required Part A (A´ for the other oligonucleotide) is the homology region shared by the target molecule and the linear molecule. Choose the way that you want to engineer your target. Often, you want to delete a section of your target. This is accomplished by replacing this section with the selectable marker. The homology regions are the about 50 bp immediately either side of the deleted section. You can delete from 0bp (i.e. make an insertion) to > 100kb. The exact sequence of the homology regions can be chosen freely, according to which position on the target molecule will be modified. But the homology arms should be unique to the target DNA and be of moderate GC-content as well as moderate complexity.
- Optional Part B (B´ for the downstream oligonucleotide): This part of the oligonucleotide allows useful sequences, such as HA-tags, Myc-tags, His-tags, or restriction sites, multiple cloning sites, site-specific recombination target sites, etc., to be incorporated into the recombinant product B and/or B´. By design, these will be incorporated into the recombinant product exactly where desired. If the introduction of such operational sequences is not needed, this piece can simply be omitted from the oligonucleotide design.
- Required Part C (C´for the downstream oligonucleotide): This piece, usually 20 to 24 nucleotides long, primes the PCR amplification of the linear molecule from the provided template. The most efficient way to design primers for modifying BACs or chromosomes using the Red/ET system is to make the electronic map of the final construct by pasting in the desired sequence into the gene to be modified. Any program like Gene Construction Kit, DNA Strider, Gene Inspector etc. will do. Based on the final construct, copying and pasting the inserted sequence plus 50 bp immediately either side of the insertion in a new file gives the electronic map of the PCR product for recombineering. Copy the upstream primer and send the sequence to synthesizer. Copy the downstream primer and make the reverse complement.
With primer design there is sometimes no choice of homology region, if for example you need to change one base pair of a particular gene. But many times you want to knock out a gene and there are no strict rules about where you must do it. Or sometimes you just want to have a marker in a particular region of a chromosome and don?t care precisely where it is. In these cases a 50 bp region that is roughly 50 % GC in content is preferred. A long stretch of a single nucleotide should better be avoided. It is a good idea to take the non-template directed extra A, that Taq polymerase puts on to the end of PCR products, into consideration. It is best to design the oligos in a way that recombination works with both the precise PCR product and the template extended-A products.
Types of plasmids
- R-plasmid -> antibiotic resistance
- F-plasmid -> conjugation
- Col-plasmids -> bacteriocins, colicins
- Degradative plasmids -> digestion of unusual substances (e.g. xylene, napthalene and camphor)
- Virulence plasmids -> bacteria gets pathogenic
Incompatibility
Plasmids are incompatible if they do not stably co-exist in the same cell. Incompatibility is based on shared replication or partitioning systems. That is, two plasmids with the same origin will be incompatible. It can be overcome by constant selection.
Most common vector plasmids
| vector name | replication origin | copy number | features | use for recombineering |
|---|---|---|---|---|
| pBR322 | pMB1 (ColE1) - unidirectional | 15-60 | original popular vector, ColE1 is naturally occuring origin, relies entirely on host proteins | targeting vector backbone, cassette amplification, subcloning |
| pUC18/19 | mutated pMB1 - unidirectional | 500-700 | easy plasmid DNA purification | not recommended |
| pACYC | p15A (ColE1-like) - unidirectional | 12-15 | compatible with other pMB1 plasmids | targeting vector backbone, cassette amplification, subcloning |
| R6K | ori gamma - unidirectional | in pir strain:15-20, in pir116 strain: ~250 | compatible with ColE1, requires Pi-Protein from pir-gene (either on plasmid or on genome), original ori had ori alpha, beta, gamma | cassette amplification, can be used to avoid any backround (explanation see "Recombineering with PCR based cassettes" last paragraph) |
| pSC101 | pSC101 - unidirectional | 4-6 | the first vector SC = Stanley Cohen, needs par locus and RepA protein for replication and partitioning, compatible with ColE1 | expression vector for recombineering and recombinase proteins, temperature sensistive replication version used |
| BAC | F plasmid oriS - unidirectional | 1-2 | very large cloning sizes, requires RepE and RepC, tight number control | ordered from genome libraries to be modified directly or to be subcloned from, internal recombination events have to be excluded |
| PAC | P1 phage plasmid origins | 3-5 | very large cloning sizes | |
| Fosmid pCC1FOS (Epicentre) | F + pMB1 - unidirectional | 1-2, inducible high copy | ~40 kb cloning size, lamdba packaging |
The author of this page thanks Madina Karimova for her information on plasmid origins.
The dimer and the mixture issue
- ideally design your recombineering experiment with middle-copy plasmids or carry them out in copy-cutter strains
- to avoid dimer formation co-electroporate the diluted high-copy plasmid DNA with the cassette-DNA
- perform restriction analysis that shows difference between original and modified plasmid
- retransform diluted DNA preparations if a mixture of modified and unmodified plasmids forms (if the mixtures shows again, it is a dimer)
- restriction digest dimers with an enzyme that cuts once in the original plasmid (in an unmodified region), religate and retransform
The mutation issue
- Use Proof-Reading-Polymerases whenever amplifying non-selectable cassettes/pieces.
- Sequence verify any PCR amplified cassette (before usage), except for bacterial promotors with bacterial selection markers, that gave the respective resistance already.
The electroporation efficiency
- Purify your PCR product with a column PCR purification kit as they help to get rid of salt and oligos, which lower electroporation and recombineering efficiency.
- Elute your PCR with water to avoid adding salt again.
The recombineering efficiency
- limit the amount of oligos you use for the PCR and purify the PCR over a column, as left over oligos compete with PCR products for single stranded DNA in the annealing step of the recombineering
The backround issue
Recombineering is often utilizes cassettes with selectable markers. Unwanted background selection is usually caused by residual intact plasmid that was used as PCR template. To reduce or eliminate recombineering background from the PCR template, you can use one of the following 5 methods or combine two of them if necessary.
Template digestion
Perform template plasmid digestion with one or more restriction enzymes with recognition sites, that are present in the plasmid backbone but absent in the drug selectable cassette to be amplified. The digested plasmid DNA needs to be checked by electrophoresis to verify efficient digestion. Furthermore, the digested DNA should be diluted to very low concentration to further reduce the background, since practically 100% digestion is always hard to achieve, especially when an inefficient restriction enzyme has to be used.
Template fragment isolation
Perform template plasmid digestion with one or more restriction enzymes with recognition sites, that are present in the plasmid backbone but absent in the drug selectable cassette to be amplified. Run the digested plasmid DNA on an agarose gel and isolate the fragment needed as template for PCR according to standard protocols.
Fragment isolation
After PCR amplification, separate the PCR-amplified targeting molecule from the template DNA by agarose gel electrophoresis. Isolate the PCR-amplicon using standard procedures. Commercially available fragment-isolation kits can be used according to manufacturer?s instructions.
DpnI digestion
The restriction enzyme DpnI has a 4-basepair recognition site (GATC), and only digests methylated DNA. Since a PCR product (i.e. linear targeting DNA) is not methylated, and template DNA usually is (upon growth in most commonly used bacterial cloning strains [dam+ strain]), DpnI only digests the template DNA and not the linear targeting molecule.
Non-replicable PCR template
Alternatively, suicide plasmids can be used as PCR template. As an example we use plasmids containing the R6K origin of replication. The pir gene encoded protein is essential to initiate the replication of the R6K plasmid. The R6K plasmid without pir gene can only replicate in a pir+-strains such as BW116, EPI100D-pir or EPI100D-pir116 cells, where the pir or the pir116 gene are present on the chromosome. Using R6K plasmids as the PCR template, consequently a pir--strain should be used for recombineering. Because the R6K plasmids cannot replicate in the recombineering strain (pir-), zero background selection is achieved without any effort of removal of the PCR template.
For successful counter selection:
- The concentration of streptomycin can be used as high as 200-500 µg/ml without killing the resistant cells.
- Low Salt LB pH 8.0 makes E.coli more sensitive to the selection.
- Take the copy number of plasmids into account: the more copies you have, the more stringent you can select. (e.g. R6K is multi-copy, BAC is 1-2 copy.)
- To perform a function test of the rpsL cassette, please don't streak too many cells on the plate. The spontaneous mutagenesis rate is 10-6. If you streak too much, you will see growth any way.
- avoid PCR amplification of large DNA fragments
- rather cut the cassette from its host plasmid and place homology arms for the cassette into the target plasmid before, by a small "adapter" cassette (for details see "practical pipeline" in the Gene Targeting Guide)
- always digest your construct with a restriction enzyme that cuts reasonably often (more than once, depending on size of plasmid 2-10 times), because missing/different bands compared to the theoretical pattern will give you information about rearrangements and mutations (if you have mutations/rearrangements you can locate the area of the problem)
- at least your final product should be digested with more than one enzymes because polymorphisms in the intronic sequences can lead to differences in the restriction patterns, that do not affect the downstream processes
- select an enzyme that gives you a clear difference between the original and the modified plasmid, best giving some bands that are specific for the original and some bands that are specific for the modified, thereby you can rule out mixtures of plasmids (see here for example)
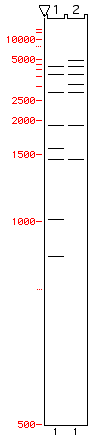{kind=link}
Storage conditions
| chemical | storage conditions |
|---|---|
| L-Arabinose (10 % in H2O, sterile filtered) | -20 °C, thaw aliquots seperately for use |
| tetracycline | -20 °C, protect from light |
| hygromycin | +4 °C, protect from light |
| kanamycin, chloramphenicol, ampicillin, blasticidin | -20 °C |
Stock and Working Conditions
| antibiotic | solvent | stock concentration mg/ml | working concentration µg/ml | for |
|---|---|---|---|---|
| Ampicillin | H2O | 100 | 50 | liquid |
| 100 | plate | |||
| Blasticidin | (Invivogen) | 10 | 30 | liquid (pH 8) |
| 40 | plate (pH 8) | |||
| Chloramphenicol | 100 % EtOH | 30 | 10 | liquid |
| 15 | plate | |||
| Gentamycin | H2O | 50 | 1 | liquid |
| 2 | plate | |||
| Hygromycin | PBS (Invitrogen) | 50 | 30 | liquid (pH 8) |
| 40 | plate (pH 8) | |||
| Kanamycin | H2O | 30 | 10 | liquid |
| 15 | plate | |||
| Spectinomycin | H2O | 100 | 60 | liquid/plate |
| Streptomycin | H2O | 50 | 100 | liquid |
| 200 | plate | |||
| Tetracycline | 75 % EtOH | 10 | 4 | liquid |
| 5 | plate |
Other antibiotics like zeocin/zeomycin, apramycin, nalidixic acid, trimethoprim lactate, puromycin and ClonNat are not used often in our lab.
Backround Information
| Rank* | Resistence | Gene | Size (bp) |
Bactericidal/ Bacteriostatic |
Comments |
|---|---|---|---|---|---|
| 1 | Chloramphenicol | chloramphenicol acetyl transferase (cat) from Tn9 | 660 | bacteriostatic | |
| 2 | Kanamycin | kanamycin phosphotransferase (aph) from Tn903 | 795 | bactericidal | |
| kanamycin and neomycin phosphotransferase II (ntpII) from Tn5 | 804 | bactericidal | select in mammalian cell culture with geneticin (G418) | ||
| 3 | Ampicillin | TEM-1 beta-lactamase (bla) from Tn3 | 861 | bactericidal | Bystander effect (satellite colonies) |
| 4 | Hygromycin-B | hygromycin phosphotransferase (hphB) from Streptomyces hygroscopicus | 1323 | bactericidal | optimal selection in low salt LB with pH 8.0 |
| 5 | Blasticidin-S | blasticidin S deaminase (BSD) from Apsergillus terreus | 399 | bactericidal | optimal selection in low salt LB with pH 8.0 |
| 6 | Zeocin** | Sh ble gene encoding bleomycin and zeocin binding protein from Streptoalloteichus hindustanus | 375 | bactericidal | optimal selection in low salt LB with pH 8.0; zeocin is light sensitive; Sh ble gene is not suitable as positive selectable marker accompanying rpsL |
| 7 | Spectinomycin | steptomycin/spectinomycin adenylyltransferase (aadA) from Enterococcus faecalis | 675 | bacteriostatic | escaping from rpsL-mediated counter selection |
| 8 | Gentamycin | gentamycin acetyltransferase and kanamycin phosphotransferase (aacA-aphD) from Tn4001 | 1440 | bactericidal | Conveying resistance to both gentamycin and kanamycin in two separate domains |
| gentamycin acetyltransferase (aacC1) from pJN105 | 534 | bactericidal | conveying resistance to gentamycin only, gentamycin is heat stable | ||
| 9 | Apramycin | apramycin acetyl transferase (aacC4) from Tn800 on R1535 | 777 | bactericidal | conveying resistance to gentamycin, kanamycin, tetracycline |
| 10 | ClonNat | nourseothricin acetyltransferase (NAT) from Streptomyces noursei | 573 | n.n. | optimal selection in low salt LB at pH 8.0 |
| 11 | Tetracycline | tetracycline efflux protein (tetA) from RP1, RP4 or Tn1721 | 1200 | bacteriostatic | tetA conveying stronger resistance than that from tet C, tetracycline is light sensitive |
| tetracycline efflux protein (tetC) from pSC101 or pBR322 | 1191 | bacteriostatic | |||
| 12 | puromycin | puromycin N-acetyltransferase (PAC) from Streptomyces alboninger | 600 | bactericidal | optimal selection in low salt LB at pH 8.0 |
| 13 | Trimethoprim | type II dihydrofolate reductase (DHFR) from Pseudomonas aeruginosa | 237 | bacteriostatic |
* Preference ranking is from group discussion based on sharpness of selection, easiness of medium and plate preparation and maintenance.
** Zeocin is inducing double strand breaks and caused problems with cloning and intramolecular recombination, therefor is not advised to use so frequently
Further Hints
E. coli is more sensitive in Low Salt LB pH 8.0. Better selection of some drug resistance need LS-LB
?Hygromycin, Blasticidin, Puromycin, Zeocin
There is cross talk of some drug selectable genes.
(avoid or titrate the concentration but depends on the strain and copy number of the plasmid)
?Apramycin ? Tetracycline, Kanamycin, Kentamycin.
?Gentamycin (1440 bp) ? Kanamycin (we use Gentamycin 534 bp)
Combination of some drugs is too sensitive to the cells
(avoid double selection or minimize the concentration)
- Kanamycin + Zeocin
- Kanamycin + Blasticidin
- Kanamycin + Spectinomycin
- probably also Blasticidin + Spectinomycin
Not all antibiotics can be used in High Throughput pipeline (liquid selection)
?Hygromycin (cells clump together against the drugs)
Some drugs are sensitive to the light
(better not order the plates from your media kitchen, but prepare fresh)
?Streptomycin, Tetracycline, Zeocin
DNA preparation: BACs and Plasmids
- Start overnight culture in a 2 ml tube. (Puncture the cap for aeration.) Incubate in Eppendorf ThermoMixer at 37 °C, 900 rpm for 14-16 hours.
- Take 30 µl as backup in a 1.5 ml tube. Centrifuge the rest of the culture at 13,200 rpm (~16,100 rcf) for 1 minute and decant the supernatant.
- Add 200 µl of P1 buffer (Qiagen, keep P1 at 4 °C because of the RNAse in the buffer)
- Mix by Eppendorf MixMate at 1,300 rpm for about 10 minutes, or until no cells are attached to the tube (or thoroughly resuspend by pipetting).
- Add 200 µl of P2 buffer (Qiagen, do not place P2 on ice, doing so will cause precipitation).
- Mix by Eppendorf MixMate at 1,300 rpm for 3-5 seconds, repeat mixing 3 times. (or invert 5 times in hands)
- Add 200 µl of buffer P3 (Qiagen, keep P3 at 4 °C) and mix by Eppendorf MixMate at 1,300 rpm for 3-5 seconds, repeat mixing 3 times. (or invert 5 times in hands)
- Gently invert the tubes three times.
- Centrifuge the white lysate at highest speed for 10-20 minutes.
- Set up new 1.5 ml tubes and add 500 µl of isopropanol.
- After centrifugation take out the supernatant with a pipette avoiding white precipitate and put it into the tube containing isopropanol. Close lids.
- Shake vigorously.
- Centrifuge at highest speed for 20 minutes. The pellet will be clear or white and will be very small and may be difficult to see.
- Decant the supernatant and invert the tubes on a piece of tissue.
- Gently add 500 µl of 70 % ethanol.
- Discard the ethanol and invert the tubes on a piece of tissue.
- You may spin the tube again and remove remaining ethanol with a pipette. Dry the inner wall of the tube with tissue or a cotton stick, avoid the pellet.
- Dry the DNA pellet at 42 °C for 10-20 minutes (ThermoMixer without shaking).
- Disolve the BAC DNA pellet in 13 µl of digestion mixture, and incubate for about 3 hours. Disolve the multi-copy plasmid DNA pellet in 12-60 μl* of TE buffer and use 4 µl for restriction analysis.
(*pSC101: 12 μl. pR6K in pir strain: 16 μl. pR6K in pir116 strain: 40 μl. p15A: 20 μl. pBR322: 40 μl. pSuperCos: 40 μl. pUC: 60 μl. pBlueScript: 60 μl.) - This protocol is also used for BAC sequencing. Then change: Disolve the DNA pellet in 8 µl of H2O, add 2 µl 10 µM sequecing primer and send to sequencing facility.
Selected Publications
The following selection of publications contains papers about recombineering strategies, targets and applications as well as the involved mechanisms, proteins and backround information of recombination (incl. endogenous factors of E. coli). The author disclaimes completeness of the paper selection. Suggestions of further important papers can be directed to the editor.
Fu, J., Bian, X., Hu, S., Wang, H., Huang, F., Seibert, P., Plaza, A., Xia, L., Müller, R., Stewart, A. F. and Zhang, Y. (2012) "Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting.” Nat Biotech, 30(5), 440 - 446.
Bird, A. W., Erler, A., Fu, J., Maresca, M., Heriche, J. K., Zhang, Y., Hyman, A. A. and Stewart, A. F. (2012). "High efficiency counterselection recombineering for site-directed mutagenesis in bacterial artificial chromosomes." Nat. Methods 9(1), 103 - 109.
Hofemeister, H., Ciotta, G., Fu, J., Seibert, P. M., Schulz, A., Maresca, M., Sarov, M., Anastassiadis, K. and Stewart, A. F. (2011). "Recombineering, transfection, Western, IP and ChIP methods for protein tagging via gene targeting or BAC transgenesis." Methods 53(4), 437 - 452n.
Justice, M. J., Siracusa, L. D. and Stewart, A. F. (2011). "Technical approaches for mouse models of human disease." Dis Model Mech 4: 305-10.
Nedelkova, M., Maresca, M., Fu, J., Rostovskaya, M., Thiede, C., Anastassiadis, K., Sarov, M. and Stewart, A. F. (2011). "Targeted isolation of cloned genomic regions by recombineering for haplotype phasing and isogenic targeting." Nucl. Acids Fes. 39(20), e137
Ciotta, G., Hofemeister, H., Maresca, M., Fu, J., Sarov, M., Anastassiadis, K. and Stewart, A. F. (2010). "Recombineering BAC transgenes for protein tagging." Methods 53(2), 113 - 119.
Fu, J., Teucher, M., Anastassiadis, K., Skarnes, W. and Stewart, A. F. (2010). "A Recombineering Pipeline to Make Conditional Targeting Constructs." Methods Enzymol 477C: 125-144.
Maresca, M., Erler, A., Fu, J., Friedrich, A., Zhang, Y. and Stewart, A. F. (2010). "Single-stranded heteroduplex intermediates in lambda Red homologous recombination." BMC Mol Biol 11: 54.
Erler, A., Wegmann, S., Elie-Caille, C., Bradshaw, C. R., Maresca, M., Seidel, R., Habermann, B., Muller, D. J. and Stewart, A. F. (2009). "Conformational adaptability of Redbeta during DNA annealing and implications for its structural relationship with Rad52." J Mol Biol 391: 586-98.
Poser, I., Sarov, M., Hutchins, J. R., Heriche, J. K., Toyoda, Y., Pozniakovsky, A., Weigl, D., Nitzsche, A., Hegemann, B., Bird, A. W., Pelletier, L., Kittler, R., Hua, S., Naumann, R., Augsburg, M., Sykora, M. M., Hofemeister, H., Zhang, Y., Nasmyth, K., White, K. P., Dietzel, S., Mechtler, K., Durbin, R., Stewart, A. F., Peters, J. M., Buchholz, F. and Hyman, A. A. (2008). "BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals." Nat Methods 5: 409-15.
Court, R., Cook, N., Saikrishnan, K. and Wigley, D. (2007). "The crystal structure of lambda-Gam protein suggests a model for RecBCD inhibition." J Mol Biol 371: 25-33.
Murphy, K. C. (2007). "The lambda Gam Protein Inhibits RecBCD Binding to dsDNA Ends." J Mol Biol 371: 19-24.
Ooi, Y. S., Warburton, P. E., Ravin, N. V. and Narayanan, K. (2007). "Recombineering linear DNA that replicate stably in E. coli." Plasmid.
Rivero-Muller, A., Lajic, S. and Huhtaniemi, I. (2007). "Assisted large fragment insertion by Red/ET-recombination (ALFIRE)--an alternative and enhanced method for large fragment recombineering." Nucleic Acids Res 35: e78.
Sawitzke, J. A., Thomason, L. C., Costantino, N., Bubunenko, M., Datta, S. and Court, D. L. (2007). "Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond." Methods Enzymol 421: 171-99.
Thomason, L. C., Costantino, N., Shaw, D. V. and Court, D. L. (2007). "Multicopy plasmid modification with phage lambda Red recombineering." Plasmid.
Velappan, N., Sblattero, D., Chasteen, L., Pavlik, P. and Bradbury, A. R. (2007). "Plasmid incompatibility: more compatible than previously thought?" Protein Eng Des Sel 20: 309-13.
Cronan, J. E. (2006). "A family of arabinose-inducible Escherichia coli expression vectors having pBR322 copy control." Plasmid 55: 152-7.
Erler, A., Maresca, M., Fu, J. and Stewart, A. F. (2006). "Recombineering reagents for improved inducible expression and selection marker re-use in Schizosaccharomyces pombe." Yeast 23: 813-23.
Perlova, O., Fu, J., Kuhlmann, S., Krug, D., Stewart, A. F., Zhang, Y. and Muller, R. (2006). "Reconstitution of the myxothiazol biosynthetic gene cluster by Red/ET recombination and heterologous expression in Myxococcus xanthus." Appl Environ Microbiol 72: 7485-94.
Sarov, M., Schneider, S., Pozniakovski, A., Roguev, A., Ernst, S., Zhang, Y., Hyman, A. A. and Stewart, A. F. (2006). "A recombineering pipeline for functional genomics applied to Caenorhabditis elegans." Nat Methods 3: 839-44.
Schnutgen, F., Stewart, A. F., von Melchner, H. and Anastassiadis, K. (2006). "Engineering embryonic stem cells with recombinase systems." Methods Enzymol 420: 100-36.
Wang, J., Sarov, M., Rientjes, J., Fu, J., Hollak, H., Kranz, H., Xie, W., Stewart, A. F. and Zhang, Y. (2006). "An improved recombineering approach by adding RecA to lambda Red recombination." Mol Biotechnol 32: 43-53.
Wu, Z., Xing, X., Bohl, C. E., Wisler, J. W., Dalton, J. T. and Bell, C. E. (2006). "Domain structure and DNA binding regions of beta protein from bacteriophage lambda." J Biol Chem 281: 25205-14.
Glaser, S., Anastassiadis, K. and Stewart, A. F. (2005). "Current issues in mouse genome engineering." Nat Genet 37: 1187-93.
Muyrers, J. P., Zhang, Y., Benes, V., Testa, G., Rientjes, J. M. and Stewart, A. F. (2004). "ET recombination: DNA engineering using homologous recombination in E. coli." Methods Mol Biol 256: 107-21.
Testa, G., Schaft, J., van der Hoeven, F., Glaser, S., Anastassiadis, K., Zhang, Y., Hermann, T., Stremmel, W. and Stewart, A. F. (2004). "A reliable lacZ expression reporter cassette for multipurpose, knockout-first alleles." Genesis 38: 151-8.
Testa, G., Vintersten, K., Zhang, Y., Benes, V., Muyrers, J. P. and Stewart, A. F. (2004). "BAC engineering for the generation of ES cell-targeting constructs and mouse transgenes." Methods Mol Biol 256: 123-39.
von Melchner, H., Stewart, A. F., Robert, L., John, G., Brigid, H., Douglas, M., Roger, P., James, T. and Michael, W. (2004). Engineering of ES Cell Genomes with Recombinase Systems. In Handbook of Stem Cells. Burlington, Academic Press: 609-622.
Costantino, N. and Court, D. L. (2003). "Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants." Proc Natl Acad Sci U S A 100: 15748-53.
Testa, G., Zhang, Y., Vintersten, K., Benes, V., Pijnappel, W. W., Chambers, I., Smith, A. J., Smith, A. G. and Stewart, A. F. (2003). "Engineering the mouse genome with bacterial artificial chromosomes to create multipurpose alleles." Nat Biotechnol 21: 443-7.
Valenzuela, D. M., Murphy, A. J., Frendewey, D., Gale, N. W., Economides, A. N., Auerbach, W., Poueymirou, W. T., Adams, N. C., Rojas, J., Yasenchak, J., Chernomorsky, R., Boucher, M., Elsasser, A. L., Esau, L., Zheng, J., Griffiths, J. A., Wang, X., Su, H., Xue, Y., Dominguez, M. G., Noguera, I., Torres, R., Macdonald, L. E., Stewart, A. F., DeChiara, T. M. and Yancopoulos, G. D. (2003). "High-throughput engineering of the mouse genome coupled with high-resolution expression analysis." Nat Biotechnol 21: 652-9.
Zhang, Y., Muyrers, J. P., Rientjes, J. and Stewart, A. F. (2003). "Phage annealing proteins promote oligonucleotide-directed mutagenesis in Escherichia coli and mouse ES cells." BMC Mol Biol 4: 1.
Casanova, E., Lemberger, T., Fehsenfeld, S., Greiner, E., Stewart, A. F. and Schütz, G. (2002). "Rapid localization of a gene within BACs and PACs." Biotechniques 32: 240-2.
Iyer, L. M., Koonin, E. V. and Aravind, L. (2002). "Classification and evolutionary history of the single-strand annealing proteins, RecT, Redbeta, ERF and RAD52." BMC Genomics 3: 8.
Shevchenko, A., Schaft, D., Roguev, A., Pijnappel, W. W. and Stewart, A. F. (2002). "Deciphering protein complexes and protein interaction networks by tandem affinity purification and mass spectrometry: analytical perspective." Mol Cell Proteomics 1: 204-12.
Copeland, N. G., Jenkins, N. A. and Court, D. L. (2001). "Recombineering: a powerful new tool for mouse functional genomics." Nat Rev Genet 2: 769-79.
Ellis, H. M., Yu, D., DiTizio, T. and Court, D. L. (2001). "High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides." Proc Natl Acad Sci U S A 98: 6742-6.
Lee, E. C., Yu, D., Martinez de Velasco, J., Tessarollo, L., Swing, D. A., Court, D. L., Jenkins, N. A. and Copeland, N. G. (2001). "A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA." Genomics 73: 56-65.
Muyrers, J. P., Zhang, Y. and Stewart, A. F. (2001). "Techniques: Recombinogenic engineering--new options for cloning and manipulating DNA." Trends Biochem Sci 26: 325-31.
Sergueev, K., Yu, D., Austin, S. and Court, D. (2001). "Cell toxicity caused by products of the p(L) operon of bacteriophage lambda." Gene 272: 227-35.
Swaminathan, S., Ellis, H. M., Waters, L. S., Yu, D., Lee, E. C., Court, D. L. and Sharan, S. K. (2001). "Rapid engineering of bacterial artificial chromosomes using oligonucleotides." Genesis 29: 14-21.
Hill, F., Benes, V., Thomasova, D., Stewart, A. F., Kafatos, F. C. and Ansorge, W. (2000). "BAC trimming: minimizing clone overlaps." Genomics 64: 111-3.
Murphy, K. C., Campellone, K. G. and Poteete, A. R. (2000). "PCR-mediated gene replacement in Escherichia coli." Gene 246: 321-30.
Muyrers, J. P., Zhang, Y., Benes, V., Testa, G., Ansorge, W. and Stewart, A. F. (2000). "Point mutation of bacterial artificial chromosomes by ET recombination." EMBO Rep 1: 239-43.
Muyrers, J. P., Zhang, Y., Buchholz, F. and Stewart, A. F. (2000). "RecE/RecT and Redalpha/Redbeta initiate double-stranded break repair by specifically interacting with their respective partners." Genes Dev 14: 1971-82.
Muyrers, J. P., Zhang, Y. and Stewart, A. F. (2000). "ET-cloning: think recombination first." Genet Eng (N Y) 22: 77-98.
Nefedov, M., Williamson, R. and Ioannou, P. A. (2000). "Insertion of disease-causing mutations in BACs by homologous recombination in Escherichia coli." Nucleic Acids Res 28: E79.
Wang, J., Chen, R. and Julin, D. A. (2000). "A single nuclease active site of the Escherichia coli RecBCD enzyme catalyzes single-stranded DNA degradation in both directions." J Biol Chem 275: 507-13.
Yu, D., Ellis, H. M., Lee, E. C., Jenkins, N. A., Copeland, N. G. and Court, D. L. (2000). "An efficient recombination system for chromosome engineering in Escherichia coli." Proc Natl Acad Sci U S A 97: 5978-83.
Zhang, Y., Muyrers, J. P., Testa, G. and Stewart, A. F. (2000). "DNA cloning by homologous recombination in Escherichia coli." Nat Biotechnol 18: 1314-7.
Angrand, P. O., Daigle, N., van der Hoeven, F., Scholer, H. R. and Stewart, A. F. (1999). "Simplified generation of targeting constructs using ET recombination." Nucleic Acids Res 27: e16.
Kuzminov, A. (1999). "Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda." Microbiol Mol Biol Rev 63: 751-813, table of contents.
Muyrers, J. P., Zhang, Y., Testa, G. and Stewart, A. F. (1999). "Rapid modification of bacterial artificial chromosomes by ET-recombination." Nucleic Acids Res 27: 1555-7.
Narayanan, K., Williamson, R., Zhang, Y., Stewart, A. F. and Ioannou, P. A. (1999). "Efficient and precise engineering of a 200 kb beta-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system." Gene Ther 6: 442-7.
Passy, S. I., Yu, X., Li, Z., Radding, C. M. and Egelman, E. H. (1999). "Rings and filaments of beta protein from bacteriophage lambda suggest a superfamily of recombination proteins." Proc Natl Acad Sci U S A 96: 4279-84.
Van Dyck, E., Stasiak, A. Z., Stasiak, A. and West, S. C. (1999). "Binding of double-strand breaks in DNA by human Rad52 protein." Nature 401: 403.
Karakousis, G., Ye, N., Li, Z., Chiu, S. K., Reddy, G. and Radding, C. M. (1998). "The beta protein of phage lambda binds preferentially to an intermediate in DNA renaturation." J Mol Biol 276: 721-31.
Li, Z., Karakousis, G., Chiu, S. K., Reddy, G. and Radding, C. M. (1998). "The beta protein of phage lambda promotes strand exchange." J Mol Biol 276: 733-44.
Zhang, Y., Buchholz, F., Muyrers, J. P. and Stewart, A. F. (1998). "A new logic for DNA engineering using recombination in Escherichia coli." Nat Genet 20: 123-8.
Mythili, E., Kumar, K. A. and Muniyappa, K. (1996). "Characterization of the DNA-binding domain of beta protein, a component of phage lambda red-pathway, by UV catalyzed cross-linking." Gene 182: 81-7.
Clark, A. J., Satin, L. and Chu, C. C. (1994). "Transcription of the Escherichia coli recE gene from a promoter in Tn5 and IS50." J Bacteriol 176: 7024-31.
Kolodner, R., Hall, S. D. and Luisi-DeLuca, C. (1994). "Homologous pairing proteins encoded by the Escherichia coli recE and recT genes." Mol Microbiol 11: 23-30.
Kowalczykowski, S. C., Dixon, D. A., Eggleston, A. K., Lauder, S. D. and Rehrauer, W. M. (1994). "Biochemistry of homologous recombination in Escherichia coli." Microbiol Rev 58: 401-65.
Hall, S. D., Kane, M. F. and Kolodner, R. D. (1993). "Identification and characterization of the Escherichia coli RecT protein, a protein encoded by the recE region that promotes renaturation of homologous single-stranded DNA." J Bacteriol 175: 277-87.
Moerschell, R. P., Das, G. and Sherman, F. (1991). "Transformation of yeast directly with synthetic oligonucleotides." Methods Enzymol 194: 362-9.
Murphy, K. C. (1991). "Lambda Gam protein inhibits the helicase and chi-stimulated recombination activities of Escherichia coli RecBCD enzyme." J Bacteriol 173: 5808-21.
Takahashi, N. and Kobayashi, I. (1990). "Evidence for the double-strand break repair model of bacteriophage lambda recombination." Proc Natl Acad Sci U S A 87: 2790-4.
Kulkarni, S. K. and Stahl, F. W. (1989). "Interaction between the sbcC gene of Escherichia coli and the gam gene of phage lambda." Genetics 123: 249-53.
Muniyappa, K. and Radding, C. M. (1986). "The homologous recombination system of phage lambda. Pairing activities of beta protein." J Biol Chem 261: 7472-8.
Lloyd, R. G. and Buckman, C. (1985). "Identification and genetic analysis of sbcC mutations in commonly used recBC sbcB strains of Escherichia coli K-12." J Bacteriol 164: 836-44.
Winans, S. C., Elledge, S. J., Krueger, J. H. and Walker, G. C. (1985). "Site-directed insertion and deletion mutagenesis with cloned fragments in Escherichia coli." J Bacteriol 161: 1219-21.
Clark, A. J., Sandler, S. J., Willis, D. K., Chu, C. C., Blanar, M. A. and Lovett, S. T. (1984). "Genes of the RecE and RecF pathways of conjugational recombination in Escherichia coli." Cold Spring Harb Symp Quant Biol 49: 453-62.
Jasin, M. and Schimmel, P. (1984). "Deletion of an essential gene in Escherichia coli by site-specific recombination with linear DNA fragments." J Bacteriol 159: 783-6.
Szostak, J. W., Orr-Weaver, T. L., Rothstein, R. J. and Stahl, F. W. (1983). "The double-strand-break repair model for recombination." Cell 33: 25-35.
Kmiec, E. and Holloman, W. K. (1981). "Beta protein of bacteriophage lambda promotes renaturation of DNA." J Biol Chem 256: 12636-9.
Wackernagel, W. (1973). "Genetic transformation in E. coli: the inhibitory role of the recBC DNase." Biochem Biophys Res Commun 51: 306-11.
Please contact us for any problems (content or function) of the Recombineering Guide.
The author also disclaims accountability for the correctness of the given information and for the content of any external links.
This guide was assembled by Madeleine Walker who thanks all the lab members helping her in gathering the information, especially Jun Fu.