Oxidative DNA "damage"
Oxidative DNA damage distributes heterogenously over the genome. AP sites are reduced in promoters and coding sequence, yet accumulate at repetitive DNA. Also 8-oxoG accumulates preferentially at repetitive DNA, particularly telomeres, short tandem repeats and potential G-quadruplex forming structures.
To understand the genomic distribution of DNA damage better, we have developped AP-Seq, a method to measure the genome wide distribution of AP sites and indirectly any base modification able to be converted specifically to AP sites, such as 8-oxoG and uracil. The original paper can be found here, the protocol here, and an associated review here.
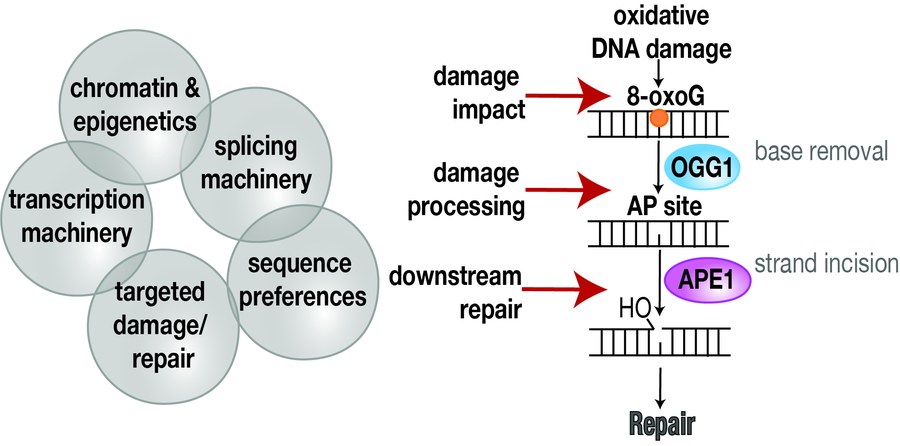
Yet, we still understand very little about the mechanisms behind the heterogenous distribution. Do the differences arise at the point of damage impact? At the step of damage processing? Downstream repair? What is influencing the specificity there? How exactly does this lead to consequences, both for the development of mutations and consequences for gene regulation. Does oxidative DNA "damage" itself function as an epigenetic mark?
To tackle this question we keep working with collaborators on optimising data analysis strategies on oxidative DNA damage distribution, as well as data analysis of related functional genomics data such as DRIP-Seq data for R-loops, G-quadruplex DNA, and data for other potential non-B-DNA structures.
A particular focus lies on how oxidative DNA damage relates to chromatin architecture and gene regulation at the nuclar pore in neural development, a collaboration with the lab of Tomohisa Toda as part of the DFG focus program Genome3 using multi-omics data like HiC, END-Seq, AP-Seq, and ChIP-Seq, and mutation data to understand such processes and their consequences for neural developmental disorders and brain tumours. Data on somatic mutagenesis, are for example available through the open Pediatric Brain Tumor Atlas to which we also contributed. For this project we are looking for a postdoc.
We are also highly interested in esophagus and stomach adenocarcinoma, whose unique mutation profiles suggest substantial involvement of oxidative DNA damage derived mutagenesis. Thus, we are collaborating with the Stange lab to generate suitable experimental models for these cancer types, helping with genetic and transcriptomic characterisation of their models, and single cell RNA-seq of organoid cultures.